IBM ve Moderna'nın çığır açan araştırması, kuantum bilgisayarların biyoloji alanındaki potansiyelini bir kez daha gözler önüne serdi. İki şirket, yapay zeka kullanmadan, 60 nükleotitlik bir mRNA zincirinin ikincil protein yapısını tahmin etmeyi başardı. Bu, şimdiye kadar bir kuantum bilgisayar üzerinde simüle edilen en uzun mRNA dizisi olma özelliğini taşıyor.
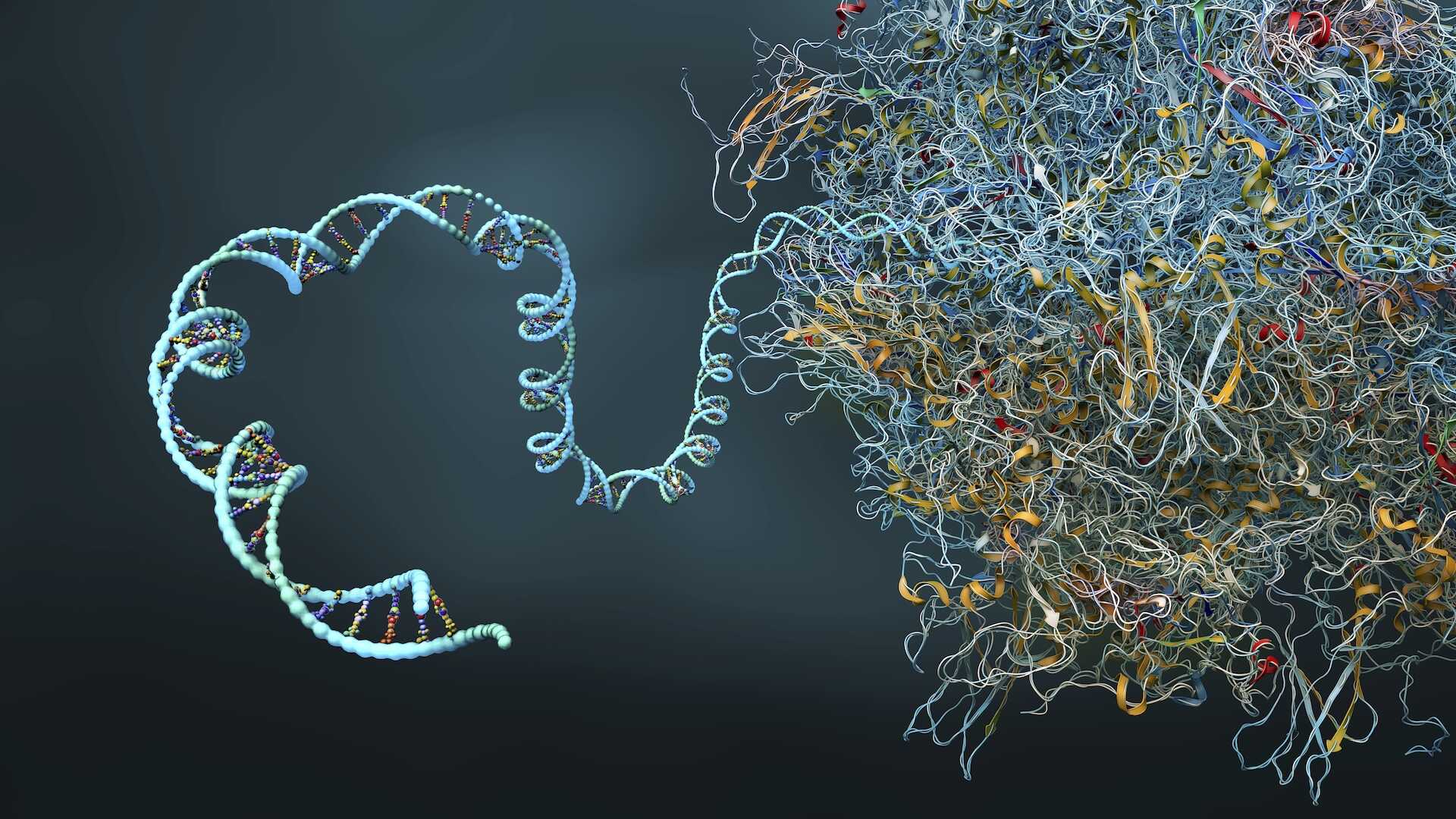
mRNA (mesajcı ribonükleik asit), hücrelerimizde genetik bilgiyi DNA'dan protein üretiminden sorumlu ribozomlara taşıyan kritik bir moleküldür. Aynı zamanda, vücudun belirli bir hastalığa karşı bağışıklık oluşturmasını sağlayan etkili aşıların geliştirilmesinde de temel bir rol oynar.
Bir proteinin doğru üç boyutlu yapısını kazanması için gereken tüm bilginin, amino asit dizisi veya 'katlanma'sında yer aldığına inanılıyor. Tek bir amino asit zincirinden oluşmasına rağmen, mRNA molekülü, belirli bir molekülün üç boyutlu şeklini oluşturan bir dizi katlanmadan oluşan ikincil bir protein yapısına sahiptir.
Her nükleotit eklendiğinde, olası katlanma permütasyonlarının sayısı katlanarak artar. Bu durum, daha uzun mRNA moleküllerinin katlanma şekillerini tahmin etme problemini, yüksek ölçeklerde çözülemez hale getirir.
IBM ve Moderna'nın bu yeni çalışması, kuantum bilgisayarların bu tür tahminler için geleneksel yöntemleri nasıl güçlendirebileceğini gösteriyor. Geleneksel olarak, bu tahminler daha çok ikili (binary) klasik bilgisayarlar ve bazı yapay zeka modelleri üzerine kuruluydu. Ancak bu yaklaşımlar, özellikle karmaşık yapıları dikkate aldığında sınırlamalara sahipti.
Son yapılan bir ön baskı çalışmasına göre, bu klasik mimarilerde çalışan algoritmalar, yüzlerce hatta binlerce nükleotitlik mRNA dizilerini işleyebiliyor, ancak bu sırada 'pseudoknot' gibi daha karmaşık özellikler dışarıda bırakılıyor. Pseudoknotlar, molekülün ikincil yapısındaki karmaşık bükülmeler ve şekillerdir ve sıradan katlanmalardan daha karmaşık iç etkileşimlere girebilirler. Bunların dışlanması, herhangi bir protein katlanma tahmin modelinin potansiyel doğruluğunu temelde sınırlar.
Bir mRNA molekülünün protein katlanmalarının en küçük ayrıntılarını anlama ve tahmin etme yeteneği, daha güçlü tahminler ve dolayısıyla daha etkili mRNA tabanlı aşılar geliştirmek için hayati önem taşır.
Bilim insanları, en güçlü süper bilgisayarların ve yapay zeka modellerinin sınırlamalarını aşmak için kuantum teknolojisi ile deneyleri güçlendirmeyi umuyor. Araştırmacılar, molekülleri modellemek için bilgisayar bitlerinin kuantum eşdeğeri olan 'qubit'leri kullanan kuantum simülasyon algoritmalarıyla çeşitli deneyler gerçekleştirdiler.
Başlangıçta, 156 qubit kapasiteli IBM R2 Heron kuantum işlem biriminin (QPU) sadece 80 qubitini kullanarak, 60 nükleotitlik bir mRNA dizisinin ikincil protein yapısını tahmin etmek için belirli bir kuantum optimizasyon algoritması uyguladılar. Çalışmaya göre, daha önceki kuantum tabanlı simülasyon modellerinde bu sınır 42 nükleotitlik bir diziydi.
Araştırmacılar ayrıca, kuantum fonksiyonlarından kaynaklanan gürültüyü (noise) yönetmek için güncel hata düzeltme tekniklerini uygulayarak deneyi ölçeklendirdiler.
Bu yeni ön baskı çalışmasında, ekip, 60 nükleotite kadar mRNA dizileri için 156 qubite kadar simüle edilmiş örnekler üzerinde deneysel paradigmanın etkinliğini geçici olarak gösterdi. Ayrıca, gürültüsüz ortamlarda aynı algoritmaları 354 qubite kadar kullanma potansiyelini gösteren ön araştırmalar da yürüttüler.
Araştırmacılar, algoritmayı çalıştırmak için kullanılan qubit sayısını artırmanın ve algoritmaları ek alt rutunler için ölçeklendirmenin daha doğru simülasyonlara ve daha uzun dizileri tahmin etme yeteneğine yol açması gerektiğini belirttiler.
Ancak, bu yöntemlerin, problem özelindeki devrelerin mevcut kuantum donanımlarına yerleştirilmesi için gelişmiş tekniklerin geliştirilmesini gerektirdiğini de eklediler. Bu da araştırmayı ilerletmek için daha iyi algoritmalar ve işlem mimarilerinin gerekeceğini gösteriyor.






